Epidermolysis bullosa is divided into four main types, differing in the level of skin at which the blisters form:
- epidermolysis bullosa simplex (EBS) - in the upper layers of the epidermis;
- borderline epidermolysis bullosa (BEB) - at the level of the light plate (lamina lucida);
- dystrophic epidermolysis bullosa (DEB) - in the upper part of the papillary dermis, below the lamina densa;
- Kindler's syndrome - different levels of blistering.
Each type is characterized by its own severity of manifestations and different combinations of mutations in different genes. The types of mutations also determine the nature of inheritance of the disease: autosomal dominant or autosomal recessive. Currently, 18 genes have been identified, mutations in which are associated with various subtypes of EB.
| Main type of BE | Main subtypes of EB | Target proteins |
| Simple BE (SBE) | Suprabasal PBE | plakophilin-1; desmoplactin; maybe others |
| Basal PBE | α6β4-integrin | |
| Borderline BE (BBE) | PoBE, Herlitz subtype | laminin-332 (laminin-5) |
| PoBE, others | laminin-332; collagen type XVII; α6β4-integrin | |
| Dystrophic EB (DEB) | dominant DBE | collagen type VII |
| recessive DBE | collagen type VII | |
| Kindler syndrome | — | kindlin-1 |
Clinical manifestations of BE
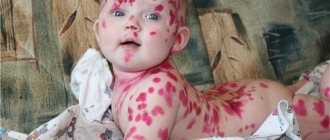
Epidermolysis bullosa manifests itself already during childbirth: the baby’s skin is injured when he passes through the birth canal. The nose, chin and heels are usually affected. In rare cases, the disease makes itself felt at 1-6 months of life. With age, depending on the type of EB, new symptoms of the disease may appear.
The main clinical manifestations of epidermolysis bullosa are blisters on the skin that appear at sites of friction, bruise, pressure, with increased body temperature, the environment, or spontaneously. Blistering can also occur on the mucous membranes of any organ. Most often the mucous membrane of the oral cavity, esophagus, intestines, genitourinary system, and eye mucosa are affected.
Timely diagnosis
Medical treatment for this disease begins with diagnosis - it makes it possible to establish an accurate diagnosis, including the subtype of epidermolysis, which makes it possible to prescribe specific treatment. In addition to the external examination, a number of specific studies are carried out:
- immunofluorescence;
- immunohistology;
They allow you to assess the level of disruption of gene structure and the depth of skin layers affected by the disease.
Also held:
- biopsy;
- genetic analysis;
- microscopy - to assess the condition of the inner layers of the skin.
It is mandatory to collect the patient's medical history - obtain information about the presence of family diseases, the duration of the disease, its manifestations and symptoms, and a comprehensive examination of all body systems, urine and blood tests.
Timely diagnosis and determination of an accurate diagnosis - a subtype of epidermolysis bullosa can provide an opportunity to improve the quality of life of a small patient and create an obstacle to the development of relapses of the disease.
Epidermolysis bullosa simplex
The modern classification of epidermolysis bullosa divides PBE into 12 subtypes. The most common subtypes of PBE are: localized subtype (formerly Weber-Cockayne type), generalized subtype (formerly Dowling-Meara or herpetiformis), generalized subtype other (formerly Koebner), epidermolysis bullosa simplex with patchy pigmentation.
The phenotype of the subtypes varies, and the blisters may appear on the hands and feet or may cover the entire body. The blisters usually heal without scarring. In rare cases, a secondary infection associated with multiple blisters can be fatal.
The most common subtype of simple EB is the localized subtype. Usually there are a large number of patients in families and the disease occurs in several generations. With this subtype, the blisters are localized on the palms and soles. At an early age they can be widespread, but with age the manifestations decrease. Clinical manifestations worsen in summer.
The most severe variant of simple EB is the generalized Dowling-Meara subtype. It is characterized by the presence of bubbles or vesicles that appear in groups. It is called EB herpetiformis because some of the lesions are similar to those of herpes simplex. The disease manifests itself at the time of birth, and the severity varies among all patients. This subtype causes widespread or confluent palmoplantar hyperkeratosis, nail dystrophy, atrophic scarring, milia, hyper- and hypopigmentation, and mucosal damage. Blistering can be severe, sometimes leading to death. This subtype can cause growth retardation and laryngeal stenosis.
Borderline epidermolysis bullosa
Borderline EB is also characterized by fragility of the skin and mucous membranes, spontaneous appearance of blisters with virtually no trauma. One sign is the formation of granulation tissue on certain parts of the body. The blisters usually heal without significant scarring.
Borderline EB is divided into two main subtypes, one of which is further subdivided into six subtypes. The main subtypes of borderline EB are: Herlitz subtype (formerly lethal), non-Herlitz subtype (formerly generalized atrophic).
The Herlitz subtype is the most severe generalized variant of borderline EB. It has a high risk of premature death. Typical symptoms include the formation of many blisters, erosions and atrophic scars of the skin, onychodystrophy leading to complete loss of the nail plates and severe scarring of the nail beds, milia, severe damage to the soft tissues in the oral cavity, enamel hypoplasia and severe dental caries. The pathognomonic symptom is abundant granulation tissue occurring symmetrically around the mouth, midface and nose, upper back, axillae, and nail folds. Possible systemic complications are severe polyetiological anemia, growth retardation, erosions and strictures of the gastrointestinal tract, damage to the mucous membranes of the upper respiratory tract and genitourinary tract, damage to the kidneys, outer membranes of the eye and, in rare cases, damage to the hands. Mortality is extremely high, especially in the first few years of life, as a result of cessation of weight gain, sepsis, pneumonia, or laryngeal and tracheal obstruction.
The non-Herlitz subtype is manifested by the formation of generalized blisters, erosions and crusts on the skin, atrophic scars, cicatricial alopecia (“male pattern”), dystrophy or loss of nails, enamel hypoplasia and caries.
Dystrophic epidermolysis bullosa
Dystrophic EB is divided into two main subtypes depending on inheritance: dominant dystrophic and recessive dystrophic (DDEB and RDEB).
Dominant dystrophic EB is clinically characterized by recurrent blistering, milia, and atrophic scarring, especially on the extremities, as well as degeneration and eventual loss of nails. In most patients, skin lesions are generalized. Extracutaneous manifestations include complications from the gastrointestinal tract.
Recessive dystrophic EB is divided into two subtypes: the severe generalized subtype (formerly Allopos-Siemens) and the generalized other subtype (formerly non-Allopos-Siemens). The recessive dystrophic severe generalized subtype is characterized by generalized formation of blisters, erosions, atrophic scars, onychodystrophy and loss of nails, pseudosyndactyly of the fingers and toes. Skin lesions are extensive and resistant to therapy. In the recessive dystrophic generalized subtype, another subtype is characterized by blisters localized on the arms, legs, knees and elbows, sometimes on the folds, on the torso.
With all subtypes of RDEB, contractures of the joints of the elbows and knees, hands and feet develop with age. Extracutaneous manifestations are often found: lesions of the gastrointestinal and genitourinary tracts, the outer membranes of the eye, chronic anemia, osteoporosis, growth retardation. Patients with RDEB have a high risk of cancer, in particular the formation of aggressive squamous cell carcinomas.
Dystrophic epidermolysis bullosa (EB) is a congenital skin disease in which the dermal-epidermal connections are disrupted, which, with the slightest physical impact, leads to detachment of the epidermis, filling of the resulting cavity with liquid and the appearance of bullous elements. The disease is caused by mutations in the COL7A1 gene, which leads to disruption of the production and distortion of the molecular structure of collagen type VII (C7). The nature of these mutations and their location determine the difference in phenotypes and the severity of the disease. Currently, there are no therapy methods with a pronounced therapeutic effect.
The development of approaches to the treatment of BE is based primarily on protein, gene and cell therapy. In a mouse model of EB, the promise of protein therapy for EB was shown, since intradermal injections of purified human C7 collagen corrected the disease phenotype and significantly increased the survival of animals.
Gene therapy approaches for BE have been carried out using various transduction vectors. In this way, in experiments, researchers were able to correct the genetic defect by transducing cDNA or the full-length C7 gene into keratinocytes or fibroblasts and achieve stable synthesis and secretion of functionally active collagen in cells.
Significant progress in solving the problem of BE has been made using cell therapy methods. Multipotent mesenchymal stromal cells of the bone marrow turned out to be suitable for this purpose, since they are capable of giving rise to functional skin cells, including keratinocytes and fibroblasts. Some researchers consider umbilical cord cells as a possible source of epithelial cells capable of epidermal reconstitution. It has also been shown that human cord blood stem cells can differentiate into keratinocytes in vitro, meaning cord blood is a good source of cells for transplantation into patients with large skin defects.
An international team of researchers recently conducted an early-phase clinical trial of allogeneic bone marrow stem cells in patients with the most severe form of EB. Despite modest initial successes, the results showed that future stem cell therapies could prolong and improve the quality of life of EB patients.
Epidermolysis bullosa (EB) is a clinically and genetically heterogeneous group of chronic skin diseases caused by mutations in a number of genes encoding structural proteins that provide dermal-epidermal connections. The National EB Registry estimates that the disease affects 20 out of 1 million US births, and although the exact number of affected individuals is unknown, it is estimated that approximately 25,000 to 50,000 Americans have EB. In most of them, the disease is mild, and its “owners” do not even seek medical help. Patients with severe forms live with constant pain, especially with lesions of the oral cavity, mucous membranes of the esophagus, rectum, and the appearance of atrophic scars on the skin in places of healed lesions can lead to scar contractures, fusion of the fingers, which is the cause of early disability, often accompanied by an unfavorable outcome In young age.
In children with a severe form of the disease, the pathology manifests itself immediately after birth or in the first years of the child’s life. Depending on the clinical picture, more than 20 forms of EB are distinguished [1], which vary significantly in the severity of manifestations and consequences. About 400 mutations in the COL7A1 gene are known to be associated with EB [2, 3], and functional defects in collagen type VII (C7), caused by dislocation of stop codons in both COL7A1 alleles, are responsible for the most serious autosomal recessive type of this disease - recessive dystrophic bullous epidermolysis (RDBE) [1].
C7 is a protein molecule that provides rigidity to the connective tissue structures of the body, such as skin, tendons and ligaments. Normally, collagen fibrils connect the top layer of skin, the epidermis, to the underlying layer, the dermis. Researchers studying the problem of EB are faced with the complexity of the structure and regulation of the expression of the COL7A1 gene. Its 9.2 kb long mRNA contains 118 exons and encodes a polypeptide with a molecular weight of 290 kDa. This protein is synthesized in skin fibroblasts and epidermal keratinocytes, secreted into the intercellular spaces, where, during the process of self-assembly, it is converted into anchor fibrils that provide strong dermo-epidermal connections [4]. The mutation of the COL7A1 gene leads to disruption of the production and distortion of the molecular structure of collagen, that is, to a situation where the connection between the epidermis and the dermis is so weak that with the slightest physical impact (pressure, abrasion, scratching), the epidermis peels off, the resulting cavity is filled with liquid and bullous formations appear .
Most of those rare patients who live into their 20s and 30s develop aggressive forms of squamous cell skin cancer. The risk of oncological transformation in RDEB increases with the age of patients; cancer can occur even at 13 years of age, and after 30, more than half of the patients expect death. It is believed that the aggressive nature of the course (high frequency of manifestation), rapid growth and early metastasis put skin cancer in first place among the causes of mortality in patients with RDEB [5, 6]. The exhausting, hopeless nature of life for EB patients, the helplessness of doctors and the despair of loved ones have prompted some countries to even consider legalizing euthanasia for newborns with the most severe forms of the disease.
Currently, there are no therapy methods with a pronounced therapeutic effect. Palliative measures come down to bandaging most of the surface of the body in order to protect the skin from possible, even the slightest mechanical influences, to prevent infection of wound areas and protect against excessive loss of tissue fluid, surgical removal of exfoliated areas of the epidermis and anesthesia of wound surfaces, provision of nutrition (mainly liquid food by mouth or using a feeding gastric tube), as well as general analgesia.
The ineffectiveness of treatment methods has prompted researchers to search for approaches and means to correct, restore, or at least compensate for the lost functions of the C7 protein in the skin of patients with EB. Initially, it was shown that type VII collagen is synthesized and secreted by basal keratinocytes [7], but later it turned out that fibroblasts in this regard are more productive, more reliable and less “fastidious” to cultivation conditions [8]. Moreover, fibroblasts have demonstrated the ability for long-term (up to 30 passages) cultivation in vitro, which allows the growth of large numbers of cells, both original and genetically modified. The identified properties of fibroblasts contributed to their further use as producers of collagen VII in the search for treatments for BE.
Attempts at protein therapy have included direct injections of recombinant C7 protein [9, 10]. Experimentally created mutant mice (Col7a1-/-) lacked collagen synthesis, as a result of which they developed all the signs of EB, leading to the death of the animals in the first week of life. Intradermal administration of recombinant human C7 protein led to the formation of collagen fibrils in the skin of animals, reduced the activity of the formation of pathological elements, and extended the life of mice to 20–25 weeks.
One promising approach to treating EB is gene therapy, which relies on methods for creating molecular constructs that can correct the functioning of mutant COL7A1. The first experiments using genetically engineered vector constructs carrying the Col7A1 gene in cell cultures [11] and laboratory animals [12] showed the technological feasibility of their use as a tool for transferring a normal gene into cells mutant for this trait.
In one of the tested experimental approaches to the treatment of EB, a study was conducted using mice hypomorphic for C7 collagen as a model of the disease (animal cells expressed no more than 10% of the normal level of collagen synthesis) with all its signs [13]. Intradermal injections of “wild type” fibroblasts with normal C7 synthesis into these animals led to the restoration of dermal-epidermal connections, and the treated skin areas showed resistance to physical influences. Untreated areas remained in an unstable state, characteristic of the classic manifestation of BE. It is assumed that further development of similar studies on a mouse model of BE may bring closer the solution to clinical problems in the treatment of this disease.
Fibroblasts isolated from a patient with EB were transfected using a lentiviral vector capable of overexpressing human C7. In an experiment on mice, wounded areas of skin were surgically created. It was shown that after intravenous administration, transfected cells migrated to damaged areas, where they were incorporated into the membrane zone of animal skin, formed typical collagen fibrillar structures and contributed to accelerated healing of skin lesions due to excessive synthesis of C7 protein. The authors consider the results as the first evidence that intravenously injected genetically modified fibroblasts can deliver C7 to wounded skin sites, and this strategy can be used in clinical practice [14].
An interesting report from Japanese researchers is that bone marrow transplantation into mice with a model disease of EB caused the formation of donor keratinocytes over the entire surface of the recipients’ skin, which was accompanied by an increase in the survival rate of laboratory animals [15]. The authors claim that both human hematopoietic and mesenchymal cells had the ability to produce full-fledged collagen. The most intriguing results of this work are the results of experiments showing that human umbilical cord blood CD34+ cells also differentiated into keratinocytes that produce components of donor skin proteins after transplantation into model animals.
Over time, there has been significant progress in technologies for using stem cells to treat skin lesions [16]. The challenge is to find a safe, reliable, sustainable and ethically appropriate source of stem cells. Multipotent mesenchymal stromal cells (MMSCs) from the bone marrow seemed to be most suitable for this, since they are able to give rise to skin cells, including keratinocytes and fibroblasts, and can also influence skin repair through the production of paracrine factors [17].
Recently, in a keratin-fibroblast co-culture model, the ability of umbilical cord epithelial cells to undergo skin-epithelial reconstitution was demonstrated [18]. Comprehensive studies of umbilical cord-derived epithelial cells have been characterized in terms of proliferative potential, telomeric length, in vitro clonogenic activity, expression of epidermal markers, and specific stem cell markers. The identified properties of umbilical cord epithelial cells inherent in human stem cells and their ability to generate specific multilayered skin epithelium, according to the authors, open the way to their clinical use in dermatological practice. It has become known that human cord blood stem cells can also differentiate into keratinocytes in vitro, meaning cord blood is a good source of cells suitable for transplantation into patients with large skin defects [19].
In recent years, specialists in the field of cell therapy have increasingly become attracted to MMSCs [20–22]. MMSCs have been described as a mononuclear population of bone marrow cells, which, when cultured in vitro, adhere well to the surface of culture plastic and acquire a fibroblast-like morphology. The International Society of Cell Therapy has established three minimum criteria characterizing MMSCs: attachment to the surface of plastic dishes during in vitro cultivation, specific expression of surface antigens (CD73+/CD90+/CD105+; CD34-/CD45-/CD11b-/CD14-/CD19-/CD79a -/ HLA-DR-) and characteristic differentiation abilities - in the osteogenic, chondrogenic and adipocyte directions [23]. A thorough study of MMSCs revealed a number of inherent properties: differentiation in vitro and in vivo into mesodermal cells [24], the ability to produce growth factors that promote proliferation, differentiation of various cells, as well as angiogenesis. The hypoimmunogenicity of MMSCs seems to be a particularly attractive property, since their transplantation does not require strict immunosuppressive measures [25, 26].
Experiments on animals showed that with local administration of MMSCs, recipients experienced restoration of damaged skin areas [17, 27]. The migration of MMSCs to sites of damage and the acquisition of the phenotype necessary for the repair of a specific tissue is regulated by a number of factors - cytokines, chemokines [28]. A recent review summarizes the current understanding of the role of stem cells in the healing of skin wounds, the ensemble of cellular factors and key molecular structures involved in the healing of skin wounds, and the publication also contains controversial information about the potential participation of populations of various skin stem cells in this process [29] .
In the context of the possibility of using MMSCs in cell therapy for BE, researchers were interested in the interaction of MMSCs with the cellular environment of the skin matrix, the influence of microenvironmental factors on them, and the ability of the matrix to ensure the differentiation of MMSCs towards skin cells [30]. The authors were able to find out that MMSCs have many common features with skin fibroblasts, and also showed that under the direct influence of the latter, both in vitro and in vivo, MMSCs acquired a fibroblast-like phenotype, which indicates the key regulatory role of intercellular interaction in the differentiation of MMSCs. This cell transition was accompanied by the activation of genes that direct the synthesis of a number of dermis-specific proteins, including type VII collagen. In animals with a model of EB, it was shown that intradermal injections of MMSCs ensured the restoration of dermal-epidermal connections. The results obtained, according to the researchers, give hope for the possibility of using MMSCs to treat patients with EB.
Among the first clinical attempts at cell therapy for BE was the treatment of a patient with one of the varieties of this pathology associated with a mutation of the LAMB3 gene [31]. The researchers introduced a retroviral vector encoding the normal LAMB3 gene into epidermal stem cells isolated from the patient. Next, “flaps” of transfected cells grown in the laboratory were applied to areas of skin after laser ablation of the patient’s epidermis. Observation lasting more than a year showed that normal protein synthesis occurred in the engrafted grafts and there were no signs of bullous lesions, i.e. stable phenotypic restoration of the skin occurred. This study showed the fundamental possibility of ex vivo gene therapy and stimulated work on the development of similar technologies for the correction of other gene mutations associated with EB. However, the fear of oncogenic consequences due to the retroviral nature of the vector and the inability to treat any large areas of the patient’s skin formed arguments in favor of searching for other means of combating this disease.
Many attempts at gene therapy for BE have relied on ex vivo cDNA transfer of the COL7A1 gene into defective keratinocytes or fibroblasts to eliminate the genetic component of EBDE. In some cases, the authors were able to achieve positive results through transgene expression, but all these approaches did not have prospects for successful clinical application.
In one of the few attempts at cell therapy for BE, researchers intradermally injected unrelated allogeneic MMSCs into two patients without immunosuppression, since the hypoimmunogenic properties of these cells are known [24]. In the absence of acute side effects in the treated skin areas, the authors noted the appearance of C7 collagen fibers, accelerated healing of wound lesions and the cessation of the emergence of new pathological elements. However, the noted improvements were temporary – from the 4th month. In both patients, symptoms of EB returned. According to the authors’ assumptions, the short duration of the therapeutic effect is due to the depletion of the pool of administered MMSCs [32].
The results of work on cell therapy, which reported the use of intradermal injections of allogeneic fibroblasts in five patients with EB [33], seem promising. The authors observed the patients for several months after the injections, and recorded an increased content of C7 collagen fibrils in the patient’s tissues, although they did not have normal morphological signs. Interesting arguments in this regard are presented by the authors of the publication in the discussion of the results obtained. The additional C7 protein, identified by various methods of analysis, is predominantly mutant; however, it would be incorrect to consider it devoid of specific functions, since the C7 protein contains many sites that ensure the stability of the skin structure. The only version in the interpretation of the results obtained by the researchers is the idea that allogeneic fibroblasts, under the influence of some paracrine effector, are capable of increasing the synthesis of fragments of the mutant C7 protein, the presence of which in excess amounts can reduce the formation of bullous lesions.
Based on the experience of previous experiments, where the authors, in experiments on mice after bone marrow transplantation, observed an increase in animal survival, C7 expression and the formation of new anchor fibrils in the skin and mucous membranes [34], an international group of medical researchers undertook the initial phase of clinical trials of donor bone marrow stem cells brain in patients with the most severe form of EB – RDEB [35]. Despite modest initial successes and losses among patients enrolled in the trials, the results showed that stem cell therapy in the future may offer hope for prolonging and improving the quality of life of patients with EB.
Recent developments in the technique of obtaining induced pluripotent stem (iPS) cells from adult somatic cells have eliminated the ethical obstacles associated with the use of embryonic stem cells and given researchers a way to grow cellular material that has the ability, under certain conditions, to differentiate into any type of cell in the body. Researchers working in the field of dermatology have taken advantage of these advances to obtain keratinocytes [36, 37] and have shown that iPS technology represents a new approach in regenerative medicine for the treatment of skin lesions, including BE. The resulting line of murine poorly differentiated keratinocytes in a reconstitutive environment in vivo was capable of complete restoration of the epidermis with the formation of skin structures such as hair follicles and sebaceous glands. The obtained iPS cells from patients with RDEB, transfected with a vector carrying the COL7A1 gene, according to the authors, can be used in systemic therapy of patients with damage to the skin and mucous membranes due to RDEB [37].
Taking into account the successes and shortcomings of previous experiments, a new technological approach has recently been proposed for the endogenous regulation of C7 collagen synthesis [38]: researchers have shown that using trans-splicing technology it is possible to correct up to one third of all mutations of the COL7A1 gene and create molecular constructs to restore the synthesis of normal full-length collagen protein in keratinocytes obtained from a patient with RDEB. In contrast to mutant keratinocytes, transfected cells acquired normal morphology, and collagen in the form of anchor fibrils was located at the base of the membrane zone in model skin equivalents. The authors of the study confidently note that the elimination of the in vitro simulated manifestations of RDBE, which they achieved using trans-splicing technology, is an important step towards its use in gene therapy in vivo.
One of the leading experts in the field of research on BE, J. Uitto (2010), notes that existing data indicate some success in solving this problem: different approaches - gene therapy, protein replacement therapy, various options for cell therapy and a number of other approaches at present have already brought research developments closer to the stage of clinical trials. Listing the advantages, inconsistencies and prospects inherent in each of the published experimental approaches, the author acknowledges that today they are all still in the early stages of scientific research, however, the totality of existing data inspires a foreboding of the approaching possibility of, if not a cure, then at least making the lives of patients suffering from bullous disease easier. epidermolysis [39].