Буллёзный эпидермолиз делится на четыре основных типа, различающихся по уровню кожи, на котором образуются пузыри:
- простой буллезный эпидермолиз (ПБЭ) — в верхних слоях эпидермиса;
- пограничный буллезный эпидермолиз (ПоБЭ) — на уровне светлой пластинки (lamina lucida);
- дистрофический буллезный эпидермолиз (ДБЭ) — в верхней части сосочкового слоя дермы, ниже плотной пластинки (lamina densa);
- синдром Киндлера — разный уровень образования пузырей.
Для каждого типа характерна своя тяжесть проявлений и различные сочетания мутаций в разных генах. Типы мутаций определяют также характер наследования заболевания: аутосомно-доминантный или аутосомно-рецессивный. В настоящее время выявлено 18 генов, мутации в которых связаны с различными подтипами БЭ.
| Основной тип БЭ | Основные подтипы БЭ | Белки-мишени |
| Простой БЭ (ПБЭ) | Супрабазальный ПБЭ | плакофилин-1; десмоплактин; возможно другие |
| Базальный ПБЭ | α6β4-интегрин | |
| Пограничный БЭ (ПоБЭ) | ПоБЭ, подтип Херлитца | ламинин-332 (ламинин-5) |
| ПоБЭ, другие | ламинин-332; коллаген XVII типа; α6β4-интегрин | |
| Дистрофический БЭ (ДБЭ) | доминантный ДБЭ | коллаген VII типа |
| рецессивный ДБЭ | коллаген VII типа | |
| Синдром Киндлера | — | киндлин-1 |
Клинические проявления БЭ
Буллёзный эпидермолиз проявляется уже во время родов: кожа ребёнка травмируется, когда он проходит через родовые пути. Обычно страдают нос, подбородок и пятки. В редких случаях болезнь дает о себе знать на 1-6 месяце жизни. С возрастом, в зависимости от типа БЭ, могут проявляться новые симптомы заболевания.
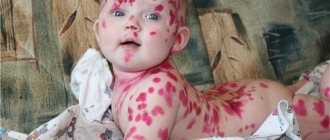
Основным клиническими проявлениями буллёзного эпидермолиза являются пузыри на коже, появляющиеся на местах трения, ушиба, давления, при повышении температуры тела, окружающей среды или спонтанно. Образование пузырей может возникать и на слизистых оболочках в любом органе. Чаще всего поражается слизистая оболочка полости рта, пищевода, кишечника, мочеполовой системы, слизистой глаз.
Своевременная диагностика
Медицинское лечение при данном заболевании начинается с диагностики — она дает возможность установить точный диагноз, включая подтип эпидермолиза, что дает возможность назначения конкретного лечения. Помимо внешнего осмотра проводится ряд специфических исследований:
- иммунофлюоресценция;
- иммуногистология;
Они позволяют оценить уровень нарушения структуры генов и глубину затронутых болезнью слоев кожи.
Также проводятся:
- биопсия;
- генетический анализ;
- микроскопия — для оценки состояния внутренних слоев кожи.
Обязателен сбор анамнеза пациента — получение информации о наличии семейных заболеваний, длительности болезни, ее проявлениях и симптомах, и комплексное обследование всех систем организма, анализы мочи и крови.
Своевременная диагностика и определение точного диагноза — подтипа буллезного эпидермолиза может дать возможность для улучшения качества жизни маленького пациента и создания препятствия для развития рецидивов заболевания.
Простой буллёзный эпидермолиз
Современная классификация буллёзного эпидермолиза делит ПБЭ на 12 подтипов. Наиболее распространёнными подтипами ПБЭ являются: локализованный подтип (ранее тип Вебера-Коккейна), генерализованный подтип (ранее Доулинга-Меары или герпетиформный), генерализованный подтип другой (ранее Кёбнера), простой буллёзный эпидермолиз с пятнистой пигментацией.
Фенотип подтипов варьируется, пузыри могут появляться на кистях рук и стопах, а могут покрывать все тело. Обычно пузыри заживают без образования рубцов. В редких случаях присоединившаяся к множественным пузырям вторичная инфекция может привести к летальному исходу.
Наиболее распространённым подтипом простого БЭ является локализованный подтип. Обычно в семьях большое количество больных и заболевание встречается в нескольких поколениях. При этом подтипе пузыри локализуется на ладонях и подошвах. В раннем возрасте могут носить распространённый характер, но с возрастом проявления уменьшаются. Клинические проявления обостряются летом.
Самый тяжёлый вариант простого БЭ – генерализованный подтип Доулинга-Меары. Он характеризуется наличием пузырей или везикул, возникающих группами. Существует название «герпетиформный простой БЭ», поскольку некоторые повреждения похожи на те, что возникают при простом герпесе. Заболевание проявляется в момент рождения, степень тяжести у всех больных варьируется. При этом подтипе отмечается распространённый или сливной ладонно-подошвенный гиперкератоз, дистрофия ногтей, атрофическое рубцевание, милии, гипер- и гипопигментация и повреждение слизистых. Образование пузырей может принимать тяжёлую форму, иногда приводящую к смерти. Этот подтип может вызывать задержку роста и стеноз гортани.
Пограничный буллёзный эпидермолиз
Пограничный БЭ также характеризуется хрупкостью кожи и слизистых оболочек, спонтанным появлением пузырей практически без травм. Одним из признаков является образование грануляционной ткани на определенных частях тела. Пузыри обычно заживают без существенных рубцов.
Пограничный тип БЭ делят на два основных подтипа, один из которых подразделяется ещё на шесть подтипов. Основные подтипы пограничного БЭ: подтип Херлитца (ранее летальный), подтип не-Херлитца (ранее генерализованный атрофический).
Подтип Херлитца наиболее тяжёлый генерализованный вариант пограничного БЭ. При нем высок риск преждевременной смерти. К типичным симптомам относятся образование множества пузырей, эрозий и атрофических рубцов кожи, ониходистрофия, приводящая к полной утрате ногтевых пластин и серьёзным рубцам ногтевых лож, милии, тяжёлое поражение мягких тканей в ротовой полости, гипоплазия эмали и тяжёлый кариес зубов. Патогномоничным симптомом является обильная грануляционная ткань, симметрично образующаяся вокруг рта, в средней части лица и вокруг носа, в верхней части спины, подмышечных впадинах и ногтевых валиках. Возможными системными осложнениями являются тяжёлая полиэтиологическая анемия, задержка роста, эрозии и стриктуры желудочно-кишечного тракта, поражение слизистых оболочек верхних дыхательных путей и мочеполового тракта, поражение почек, наружных оболочек глаза и в редких случаях поражение кистей рук. Смертность крайне высока, особенно в первые несколько лет жизни, в результате прекращения прибавки в весе, сепсиса, пневмонии или обструкции гортани и трахеи.
Подтип не-Херлитца проявляется образованием генерализованных пузырей, эрозий и корок на коже, атрофических рубцов, рубцовой алопеции («по мужскому типу»), дистрофией или потерей ногтей, гипоплазией эмали и кариесом.
Дистрофичный буллезный эпидермолиз
Дистрофический БЭ делится на два главных подтипа в зависимости от наследования: доминантный дистрофический и рецессивный дистрофический (ДДБЭ и РДБЭ).
Доминантный дистрофический БЭ клинически характеризуется рецидивирующим образованием пузырей, милиями и атрофическим рубцеванием, особенно на конечностях, а также дистрофией и, в конечном итоге, утратой ногтей. У большинства пациентов поражение кожи является генерализованным. К внекожным проявлениям относятся осложнениями со стороны желудочно-кишечного тракта.
Рецессивный дистрофический БЭ делится на два подтипа: тяжёлый генерализованный подтип (ранее Аллопо-Сименса) и генерализованный другой подтип (ранее не-Аллопо-Сименса). Рецессивный дистрофический тяжёлый генерализованный подтип характеризуется генерализованным образованием пузырей, эрозиями, атрофическими рубцами, ониходистрофией и утратой ногтей, псевдосиндактилией пальцев рук и ног. Поражение кожи носит обширный и устойчивый к терапии характер. При рецессивном дистрофическом генерализованном другом подтипе характеризуется пузыри локализуются на руках, ногах, коленях и локтях, иногда на сгибах, на туловище.
При всех подтипах РДБЭ с возрастом развиваются контрактуры суставов локтей и коленей, кистей и стоп. Часто встречаются внекожные проявления: поражения желудочно-кишечного и мочеполового трактов, внешних оболочек глаза, хроническую анемию, остеопороз, задержку роста. У пациентов с РДБЭ высокий риск онкологических заболеваний, в частности образования агрессивных плоскоклеточных карцином.
Дистрофический буллезный эпидермолиз (БЭ) является врожденным заболеванием кожи, при котором нарушены дермо-эпидермальные связи, что при малейшем физическом воздействии приводит к отслоению эпидермиса, заполнению образовавшейся полости жидкостью и появлению буллезных элементов. Заболевание обусловлено мутациями в гене COL7A1, что приводит к нарушению выработки и искажению молекулярной структуры коллагена типа VII (С7). Характер этих мутаций и их локализация определяют различие фенотипов и тяжесть протекания болезни. В настоящее время не существует методов терапии с выраженным лечебным эффектом.
Разработка подходов к лечению БЭ основывается преимущественно на белковой, генной и клеточной терапии. На мышиной модели БЭ была показана перспективность белковой терапии БЭ, поскольку внутрикожные инъекции очищенного коллагена C7 человека исправляли болезненный фенотип и существенно повышали выживаемость животных.
Подходы к генной терапии БЭ осуществлялись с использованием различных векторов трансдукции. Таким путем исследователям удавалось в экспериментах с помощью трансдукции кДНК либо полноразмерного гена С7 в кератиноциты или фибробласты исправлять генетический дефект, добиваться стабильного синтеза и секреции в клетках функционально активного коллагена.
Существенный прогресс в решении проблемы БЭ наметился при использовании методов клеточной терапии. Пригодными для этого оказались мультипотентные мезенхимные стромальные клетки костного мозга, поскольку способны давать начало функциональным клеткам кожи, включая кератиноциты и фибробласты. Некоторые исследователи рассматривают клетки пуповины как возможный источник эпителиоцитов, способных к эпидермальной реконституции. Показано также, что стволовые клетки пуповинной крови человека in vitro могут дифференцироваться в кератиноциты, то есть пуповинная кровь представляет собой хороший источник получения клеток для трансплантации пациентам с большими дефектами кожи.
Международная группа исследователей недавно осуществила начальную фазу клинических испытаний аллогенных стволовых клеток костного мозга на больных с наиболее тяжелой формой БЭ. Несмотря на скромные первые успехи, результаты показали, что терапия с использованием стволовых клеток в будущем может продлить и улучшить качество жизни больных БЭ.
Буллёзный эпидермолиз (БЭ) представляет собой клинически и генетически гетерогенную группу хронических заболеваний кожи, обусловленных мутациями ряда генов, кодирующих структурные белки, которые обеспечивают дермо-эпидермальные связи. По оценкам Национального Регистра БЭ, заболевание встречается у 20 младенцев из 1 млн новорожденных в США, и, хотя точное количество больных неизвестно, предполагается, что приблизительно 25–50 тыс. американцев страдают БЭ. У большинства из них болезнь протекает в легкой форме, и ее «обладатели» даже не обращаются за медицинской помощью. Пациенты с тяжелыми формами живут с постоянной болью, особенно при поражениях полости рта, слизистых пищевода, прямой кишки, а возникновение атрофических рубцов на коже в местах заживших поражений может приводить к рубцовым контрактурам, сращению пальцев, что является причиной ранней инвалидности, зачастую сопровождается неблагоприятным исходом в молодом возрасте.
У детей с тяжелой формой заболевания патология проявляется сразу после рождения или в первые годы жизни ребенка. В зависимости от клинической картины различают более 20 форм БЭ [1], значительно различающихся по тяжести проявлений и последствий. Известно около 400 мутаций в гене COL7A1, сопряженных с БЭ [2, 3], а функциональные дефекты коллагена типа VII (С7), вызванные дислокацией терминирующих кодонов в обоих аллелях COL7A1, ответственны за наиболее серьезный аутосомно-рецессивный тип этого заболевания – рецессивный дистрофический буллезный эпидермолиз (РДБЭ) [1].
С7 представляет собой белковые молекулы, обеспечивающие жесткость соединительно-тканных структур организма, таких как кожа, сухожилия и связки. В норме фибриллы коллагена связывают верхний слой кожи – эпидермис – с нижележащим слоем – дермой. Исследователи, занимающиеся проблемой БЭ, сталкиваются со сложностью структуры и регуляции экспрессии гена COL7A1. Его мРНК длиной 9,2 kb содержит 118 экзонов и кодирует полипептид с молекулярной массой 290 kDa. Этот белок синтезируется в кожных фибробластах и эпидермальных кератиноцитах, секретируется в межклеточные пространства, где в процессе самосборки преобразуется в якорные фибриллы, обеспечивающие прочные дермо-эпидермальные связи [4]. Мутация гена COL7A1 приводит к нарушению выработки и искажению молекулярной структуры коллагена, то есть к ситуации, когда связь эпидермиса с дермой настолько слаба, что при малейшем физическом воздействии (надавливание, потертость, расчесывание) происходит отслоение эпидермиса, заполнение образовавшейся полости жидкостью и появлению буллезных образований.
У большинства из тех редких больных, кто доживает до 20–30-летнего возраста развиваются агрессивные формы плоскоклеточного рака кожи. Риск онкологической трансформации при РДБЭ увеличивается с возрастом пациентов, рак может возникнуть даже в 13 лет, а после 30 – летальный исход ожидает более половины больных. Считается, что агрессивный характер течения (высокая частота манифестации), быстрый рост и раннее метастазирование – ставят рак кожи на первое место среди причин летальности больных РДБЭ [5, 6]. Изматывающий, безнадежный характер жизни больных БЭ, беспомощность медиков и отчаяние близких побудили некоторые страны даже к рассмотрению легализации эвтаназии для новорожденных с наиболее тяжелыми формами заболевания.
В настоящее время не существует методов терапии с выраженным лечебным эффектом. Паллиативные мероприятия сводятся к бинтованию большей части поверхности тела с целью защиты кожи от возможных, даже легчайших механических воздействий, для предотвращения инфицирования раневых участков и защиты от чрезмерной потери тканевой жидкости, хирургическому удалению отслоившихся участков эпидермиса и обезболиванию раневых поверхностей, обеспечению питанием (в основном жидкой пищей через рот либо с помощью питательного желудочного зонда), а также общей аналгезии.
Неэффективность методов лечения побудила исследователей к поискам подходов и средств исправления, восстановления или хотя бы компенсации утраченных функций С7 белка в коже больных БЭ. Поначалу было показано, что коллаген VII типа синтезируется и секретируется базальными кератиноцитами [7], но позднее выяснилось, что фибробласты в данном отношении более продуктивны, более надежны и менее «привередливы» к условиям культивирования [8]. Более того, фибробласты проявили способность к длительному (до 30 пассажей) культивированию in vitro, что позволяет наращивать большие количества клеток как исходных, так и генетически модифицированных. Выявленные свойства фибробластов способствовали дальнейшему их использованию как продуцентов коллагена VII в поисках средств лечения БЭ.
Предпринятые попытки белковой терапии включали прямые инъекции рекомбинантного белка С7 [9, 10]. У экспериментально созданных мутантных мышей (Col7a1-/-) отсутствовал синтез коллагена, вследствие чего у них возникали все признаки БЭ, приводившие к смерти животных на первой неделе жизни. Внутрикожное введение рекомбинантного C7 белка человека приводило к формированию коллагеновых фибрилл в коже животных, снижало активность образования патологических элементов и продлевало жизнь мышей до 20–25 нед.
Одним из многообещающих подходов к лечению БЭ является генная терапия, которая опирается на методы создания молекулярных конструкций, способных исправить работу мутантного COL7A1. Первые эксперименты с применением в культурах клеток [11] и на лабораторных животных [12] генноинженерных векторных конструкций, несущих ген Col7A1, показали технологическую возможность их использования в качестве инструмента переноса нормального гена в клетки, мутантные по этому признаку.
В одном из опробованных экспериментальных подходов к лечению БЭ было проведено исследование с использованием гипоморфных по С7 коллагену мышей как модели заболевания (клетки животных экспрессировали не более 10% от нормального уровня синтеза коллагена) со всеми его признаками [13]. Внутрикожные инъекции этим животным фибробластов «дикого типа» с нормальным синтезом С7 приводили к восстановлению дермо-эпидермальных связей, обработанные участки кожи проявляли устойчивость к физическим воздействиям. Необработанные участки оставались в неустойчивом состоянии, характерном для классического проявления БЭ. Предполагают, что дальнейшее развитие подобных исследований на мышиной модели БЭ может приблизить решение клинических проблем в лечении этого заболевания.
С помощью лентивирусного вектора, способного к суперэкспрессии человеческого С7, были трансфицированы фибробласты, выделенные от больного БЭ. В эксперименте на мышах хирургическим путем создавали раневые участки кожи. Было показано, что после внутривенного введения трансфицированные клетки мигрировали к поврежденным участкам, где инкорпорировались в мембранную зону кожи животных, формировали типичные коллагеновые фибриллярные структуры и способствовали ускоренному заживлению кожных поражений благодаря избыточному синтезу С7 белка. Авторы рассматривают полученные результаты как первое доказательство того, что внутривенно введенные генетически модифицированные фибробласты могут доставлять C7 к раневым участкам кожи, и такая стратегия может быть использована в клинической практике [14].
Интересно сообщение японских исследователей, повествующее о том, что трансплантация костного мозга мышам с модельным заболеванием БЭ вызывала образование донорских кератиноцитов по всей поверхности кожи реципиентов, что сопровождалось повышением выживаемости лабораторных животных [15]. Авторы утверждают, что способностью продуцировать полноценный коллаген обладали как гемопоэтические, так и мезенхимальные клетки человека. Наиболее интригующими в данной работе выглядят результаты экспериментов, показавших, что CD34+ клетки пуповинной крови человека также дифференцировались в кератиноциты, продуцирующие компоненты донорских кожных белков после трансплантации модельным животным.
Со временем наметился существенный прогресс в технологиях применения стволовых клеток для лечения повреждений кожи [16]. Задача состоит в том, чтобы найти безопасный, надежный, устойчивый и этически подходящий источник стволовых клеток. Для этого, казалось, наиболее пригодны мультипотентные мезенхимные стромальные клетки (ММСК) костного мозга, поскольку они способны давать начало клеткам кожи, включая кератиноциты и фибробласты, а также могут влиять на восстановление кожи путем продукции паракринных факторов [17].
Недавно в модели кератин-фибробластного cокультивирования была показана способность эпителиальных клеток пуповины к кожно-эпителиальной реконституции [18]. Всесторонние исследования выделенных из пуповины эпителиальных клеток охарактеризованы с точки зрения пролиферативного потенциала, длины теломерных участков, клоногенной активности in vitro, экспрессии эпидермальных маркеров и специфичных маркеров стволовых клеток. Выявленные свойства эпителиальных клеток пуповины, присущие стволовым клеткам человека, и их способность генерировать специфический многослойный кожный эпителий, по мнению авторов, открывают путь к их клиническому применению в практике дерматологии. Стало известно, что стволовые клетки пуповинной крови человека также могут in vitro дифференцироваться в кератиноциты, то есть пуповинная кровь представляет собой хороший источник клеток, пригодных для трансплантации пациентам с большими дефектами кожи [19].
В последние годы специалистов в области клеточной терапии все больше стали привлекать ММСК [20–22]. ММСК были описаны как мононуклеарная популяция костномозговых клеток, которые при культивировании in vitro хорошо адгезируются к поверхности культурального пластика и приобретают фибробластоподобную морфологию. Международным Сообществом Клеточной Терапии было установлено три минимальных критерия, характеризующих ММСК: прикрепление к поверхности пластиковой посуды при культивировании in vitro, специфическая экспрессии поверхностных антигенов (CD73+/CD90+/ CD105+; CD34-/CD45-/CD11b-/CD14-/CD19-/CD79a-/ HLA-DR-) и характерные способности к дифференцировке – в остеогенном, хондрогенном и адипоцитарном направлениях [23]. Тщательное изучение ММСК выявило ряд присущих им свойств: дифференцировка in vitro и in vivo в клетки мезодермального ряда [24], способность продуцировать факторы роста, способствующие пролиферации, дифференцировке различных клеток, а также ангиогенезу. Гипоиммуногенность ММСК выглядит особенно привлекательным свойством, поскольку при их трансплантации не требуется проведения жестких иммунодепрессивных мероприятий [25, 26].
В экспериментах на животных было показано, что при локальном введении ММСК у реципиентов наблюдалось восстановление поврежденных участков кожи [17, 27]. Миграция ММСК к местам повреждений и приобретение фенотипа, необходимого для репарации конкретной ткани, регулируется рядом факторов – цитокинов, хемокинов [28]. В недавнем обзоре суммированы данные о современном понимании роли стволовых клеток в заживлении кожных повреждений, ансамбле клеточных факторов и ключевых молекулярных структур, причастных к заживлению кожных ран, публикация содержит также дискуссионные сведения о потенциально возможном участии популяций различных кожных стволовых клеток в этом процессе [29].
В контексте возможности использования ММСК в клеточной терапии БЭ исследователей интересовали вопросы взаимодействия ММСК с клеточным окружением кожного матрикса, влиянием на них факторов микроокружения и способности матрикса обеспечивать дифференцировку ММСК в направлении клеток кожи [30]. Авторам удалось выяснить, что MМCК имеют много общих черт с кожными фибробластами, а также показать, что под непосредственным влиянием последних как in vitro, так и in vivo ММСК приобретали фибробластоподобный фенотип, что свидетельствует о ключевой регуляторной роли межклеточного взаимодействия в дифференцировке ММСК. Такой переход клеток сопровождался активацией генов, направляющих синтез ряда специфицеских для дермы белков, в том числе коллагена VII типа. На животных с моделью БЭ было показано, что внутрикожные инъекции ММСК обеспечивали восстановление дермо-эпидермальных связей. Полученные результаты, по мнению исследователей, дают надежду на возможность использования ММСК для лечения пациентов с БЭ.
Среди первых клинических попыток клеточной терапии БЭ было лечение пациента с одной из разновидностей этой патологии, связанной с мутацией гена LAMB3 [31]. В изолированные от больного эпидермальные стволовые клетки исследователи ввели ретровирусный вектор, кодирующий нормальный LAMB3 ген. Далее, выращенные в лаборатории «лоскуты» трансфицированных клеток накладывали на участки кожи после лазерной абляции эпидермиса пациента. Наблюдение продолжительностью более года показало, что в приживленных трансплантатах происходил синтез нормального белка, отсутствовали признаки буллезного поражения, т.е. наступило устойчивое фенотипическое восстановление кожи.Данное исследование показало принципиальную возможность ex vivo генной терапии и стимулировало работы по развитию подобных технологий для коррекции и других генных мутаций, связанных с БЭ. Однако опасение онкогенных последствий в связи с ретровирусной природой вектора, невозможность обработки сколько-нибудь обширных участков кожи пациента составили аргументы в пользу поиска иных средств борьбы с этим заболеванием.
Множество попыток генной терапии БЭ основывались на ex vivo переносе кДНК COL7A1 гена в дефектные кератиноциты или фибробласты, чтобы устранить генетическую составляющую РДБЭ. В некоторых случаях авторам удавалось добиться позитивных результатов за счет экспрессии трансгена, однако все эти подходы не имели перспектив успешного клинического применения.
В одной из немногочисленных попыток клеточной терапии БЭ исследователи внутрикожно вводили неродственные аллогенные ММСК двум пациентам без иммуносуппрессии, поскольку известны гипоиммуногенные свойства этих клеток [24]. При отсутствии острых побочных эффектов в обработанных участках кожи авторы отмечали появление С7 коллагеновых волокон, ускорение заживления раневых поражений и прекращение возникновения новых патологических элементов. Однако отмеченные улучшения носили временный характер – с 4-го мес. у обоих пациентов симптомы БЭ возобновились. По предположениям авторов, кратковременность лечебного эффекта обусловлена истощением пула введенных ММСК [32].
Перспективными представляются результаты работы по клеточной терапии, где сообщалось о применении внутрикожных инъекций аллогенных фибробластов пяти пациентам с БЭ [33]. Авторы наблюдали больных несколько месяцев после выполнения инъекций, при этом регистрировали в тканях пациентов повышенное содержание С7-коллагеновых фибрилл, хотя и не имевших нормальных морфологических признаков. Интересные рассуждения на этот счет изложены авторами публикации в обсуждении полученных результатов. Дополнительный С7-белок, выявленный разными методами анализа, является преимущественно мутантным, тем не менее, неправильно было бы считать его лишенным специфических функций, поскольку С7 белок содержит множество участков обеспечения стабильности кожной структуры. Единственной версией в интерпретации полученных результатов исследователи выдвигают представление о том, что аллогенные фибробласты под действием какого-то паракринного эффектора способны повышать синтеза фрагментов мутантного С7-белка, присутствие которого в избыточном количестве может уменьшить формирование буллезных поражений.
По опыту предыдущих экспериментов, где авторы в опытах на мышах после трансплантации костного мозга наблюдали повышение выживаемости животных, экспрессию С7 и формирование новых якорных фибрилл в коже и слизистых [34], международная группа исследователей в области медицины предприняла начальную фазу клинических испытаний донорских стволовых клеток костного мозга на больных с наиболее тяжелой формой БЭ – РДБЭ [35]. Не смотря на скромные первые успехи и потери из числа включенных в испытания пациентов, результаты показали, что терапия с использованием стволовых клеток в будущем сможет дать надежду на продление и улучшение качества жизни больных БЭ.
Развитая в последние годы техника получения из соматических клеток взрослого индивида индуцированных плюрипотентных стволовых клеток (iPS) устранила этические препятствия, связанные с использованием эмбриональных стволовых клеток, и дала исследователям способ выращивания клеточного материала, обладающего способностью в определенных условиях дифференцироваться в любой тип клеток организма. Исследователи, работающие в области дерматологии, воспользовались этими достижениями для получения кератиноцитов [36, 37] и показали, что технология iPS представляет собой в регенеративной медицине новый подход к лечению кожных поражений, в том числе БЭ. Полученная линия мышиных малодифференцированных кератиноцитов в реконститутивном окружении in vivo способна была к полному восстановлению эпидермиса с образованием таких кожных структур, как волосяные фолликулы и сальные железы. Полученные iPS клетки от больных РДБЭ, трансфицированные вектором, несущим COL7A1 ген, по мнению авторов, могут быть использованы в системной терапии пациентов с повреждениями кожи и слизистых при РДБЭ [37].
С учетом успехов и недостатков предыдущих экспериментов для эндогенной регуляции синтеза С7 коллагена недавно был предложен новый технологический подход [38]: исследователи показали, что путем технологии транс-сплайсинга можно корректировать до одной трети всех мутаций COL7A1 гена и создавать молекулярные конструкции для восстановления синтеза нормального полноразмерного белка коллагена в кератиноцитах, полученных от больного РДБЭ. В отличие от мутантных кератиноцитов трансфицированные клетки приобретали нормальную морфологию, а коллаген в виде якорных фибрилл располагался в основании мембранной зоны в модельных кожных эквивалентах. Авторы исследования с уверенностью отмечают, что достигнутое ими с помощью технологии транс-сплайсинга устранение смоделированных in vitro проявлений РДБЭ является важным шагом на пути применения его в генной терапии in vivo.
Один из ведущих специалистов в области исследований по БЭ J. Uitto (2010) отмечает, что существующие данные свидетельствуют о некоторых успехах в решении этой проблемы: разные подходы – генная терапия, заместительная белковая терапия, различные варианты клеточной терапии и ряд других подходов в настоящее время уже приблизили исследовательские разработки к стадии клинических испытаний. Перечисляя достоинства, противоречивость и перспективы, присущие каждому из опубликованных экспериментальных подходов, автор признает, что все они сегодня еще находятся на ранних стадиях научного поиска, однако совокупность существующих данных внушает предчувствие приближающейся возможности если не излечения, то хотя бы облегчения жизни пациентов, страдающих буллезным эпидермолизом [39].